Introduction
The The renin-angiotensin-aldosterone system (RAAS) controls blood pressure while maintaining bodily fluids balance. Its principal active hormone, angiotensin II (Ang II), is generated through a sequential process involving the breakdown of peptides originating from the initial angiotensinogen molecule. Ang II interacts with distinct receptors, setting off diverse arrays of physiological responses that affect various body systems, such as the cerebrum, cardiac, kidneys, blood vessels, and the body’s immune system. However, the major function of the RAAS lies in upholding vascular homeostasis and safeguarding optimal body fluid levels [1,2]. Ang II activities include stimulation of two kinds of receptors, namely AT1R and AT2R. While AT2R is largely expressed during fetal development and becomes more active in pathological situations, AT1R is widely distributed in adult tissues. The major biological effects of Ang II, such as blood pressure control, salt and water retention, hormone secretion, and renal function, as well as local impacts of Ang II on cellular growth, movement, and extracellular matrix synthesis, are mediated through binding to AT1R. Studies showed that Ang II interaction with AT2R is well known for counteracting the effects mediated by AT1R [3]. In addition to the well-known systemic effects of RAAS, various organs and tissues contain their own localized RAAS which mediate a wide range of cellular processes, including tissue inflammation, fibrogenesis, cell proliferation, differentiation, apoptosis, and the production of reactive oxygen species (ROS) [4,5]. It is crucial to remember that systemic and local RAASs function in a complementary and coordinated manner. Studies have revealed that key components of the RAAS are present in the normal liver, and their expression change in response to liver injuries [6]. Hepatocytes, bile duct cells, hepatic stellate cells, myofibroblasts, Kupffer cells, and vascular endothelial cells are among liver cell types that have the AT1R receptor, Ang II largely exerts its effects on the liver through this receptor. Additionally, some studies have noted the presence of the AT2R gene in liver tissue, indicating that AT2R might exert protective influences against hepatic fibrosis [6,7]. The AT2R mediates several tissue-protective actions in the pathophysiological setting, including anti-inflammation, immune modulation, anti-fibrosis, inhibition of sympathetic outflow, anti-apoptosis, and anti-neurodegeneration [8,9]. Many of these activities converge in the setting of heart failure in a well-coordinated manner. For instance, AT2R stimulation performs anti-inflammatory activity by reducing cytokine production and anti-fibrotic effect by inhibiting TGF-β formation in rats with heart failure induced by cardiac infarction; therefore, improving peri-infarct remodeling and enhancing cardiovascular performance [10,11]. Moreover, the advantageous effects of stimulating AT2R have been observed in various other disease models, such as cardiovascular conditions, complications related to diabetes, autoimmune disorders, neurological ailments, and others. These findings have been associated with improved overall health outcomes [12]. Aside from its primary physiological roles, an imbalance within the RAAS can significantly contribute to the onset of health issues such as high blood pressure, cardiac hypertrophy, and heart attack. Consequently, drugs designed to hinder the production or actions of Ang II have proven to be highly beneficial in the field of cardiovascular therapy. Blockades of the excessive activation of the RAAS through various medications, such as angiotensin receptor blockers (ARBs), significantly affect the management of conditions such as high blood pressure, congestive heart failure, and kidney disorders [13]. Azilsartan, an innovative ARB, exhibits superior efficacy in lowering blood pressure compared to its counterparts within the same class. This heightened effectiveness stems from its increased binding affinity to AT1 receptors and slow dissociation rate from them [14]. Previous research demonstrates that azilsartan exhibits various pharmacological effects and multifaceted health advantages related to endothelial failure, stroke, breast tumors, inadequate kidney supply, and pulmonary damage [15,16]. Excessive fat intake triggers the hepatoprotective effect of azilsartan on nonalcoholic liver disease, as revealed by a previous study [17]. Consequently, the present research aimed to assess the potential defensive properties of two different concentrations of azilsartan on the liver and heart of rats challenged with ethanol. Additionally, to gain a deeper understanding of the way these enzymes function, their activity was assessed, and their interactions at the active site were analyzed through molecular docking studies involving target proteins. This approach enhanced our comprehension of the mechanisms of action of these enzymes.
Materials and Methods
Thirty-two adult male Wistar Albino rats, ranging in weight from 180 to 230 g, were sourced from the animal house at the University of Sulaimani. They were housed in adequately ventilated plastic cages, maintaining a temperature of 25 °C and a humidity level of 55%, with a 12-hour light-dark cycle. The rats had ad libitum access to standard laboratory chow and water. Before the experiment began, a one-week acclimatization period was observed. The research procedures adhered to the guidelines outlined by the Institutional Animal Ethics Committee and were approved by the Ethical Committee of the University of Sulaimani, specifically its College of Pharmacy (Certificate No. PH34-21 on 20th October 2023). The study was carried out under the 1998 standards of the Canadian Council on Animal Care. The animals were randomly divided into four groups. Ethanol dosages, administration routes, and treatment groups were determined based on prior research [18,19].
The following groups each have eight rats:
• Negative Control group: Over a period of 14 days, orally via a gavage tube 1 mL of distilled water was given.
• Positive Control group: For 14 days, orally through a gavage tube animals were given 1 mL of distilled water before receiving 1 mL of 80% ethanol orally 2 h later on day 14th.
• Azilsartan treatment groups (16 animals, 8 per dose): Over 14 days, 1 mg/kg and 10 mg/kg were used orally via a gavage tube daily.
On 14th day of the experiment, all animals underwent a 24-hour fasting period during which they only had unrestricted access to water. Following this fast, with the exception of the negative control group, each animal received an oral dose of 1 ml of 80% ethanol 2 h subsequent to the initial treatment administration, and scarification was performed 1 h later.
Estimation of atherogenic indices:
The indices related to atherosclerosis were computed in the following manner: [20]
Cardiac Risk Ratio (CRR) =TC/HDLC
Atherogenic Coefficient (AC) = (TC – HDLC)/HDLC
Atherogenic Index of Plasma (AIP) = log (TG/HDLC).
Biochemical tests
At the end of the study on day 14, blood samples were collected via cardiac puncture. The blood was centrifuged, and the serum was separated and used to assess serum lipids profile, liver enzymes, cardiac risk ratio, atherogenic index of plasma, hematological markers, TNF-α, MDA, and TAOC, by using ELISA kit (Bioassay technology laboratory, UK) according to the manufacturer's instructions. C-reactive protein (CRP) levels were measured using an enzyme-linked immunosorbent test kit (Elabscience, Houston, TX, USA) as directed by the manufacturer.
Molecular docking study
Azilsartan was received from PubChem, saved as an Structural data file (SDF), and then converted to a Protein Data Bank (PDB) file with Open Babel. The ligand energy was reduced using the Merck Molecular Force Field 94 (MMFF94) force field method, and the scaled structure was converted to Protein Data Bank, Partial Charge (Q) & Atom Type (T) (PDBQT) format using PyRx 0.8 before molecular docking analysis. The six proteins' crystal structures were obtained from the PDB (PDB IDs: 7T83, 7DOV, 1B09 , 1Z9H, 3TCM, and 1AAM) [21,22]. The Discoverystudio2021 client was used to remove water molecules, heteroatoms, co-crystallized ligands, and all protein chains except for chain A. Autodock-Tool-1.5.6 29 was used to apply the polar hydrogens and Kollman charges. PyRx was used to convert the PDB file into PDBQT format. Finally, the PyRx docking tool (Python Prescription 0.8) was used to dock Azilsartan with previously synthesized proteins. The binding site is selected based on the co-crystallized ligand of the target proteins. PyRx affinity scores (in kcal/mol) for the chemical were gathered and analyzed using the free energy binding theory (more negative values indicate stronger binding affinity). University of California, San Francisco Chimera (UCSF) Chimera 1.15 and the Discoverystudio2021 client were used to show immobilization (posture) configurations and receptor-ligand interactions at the molecular level [23].
Statistical evaluation
GraphPad Prism 8 was used to statistically analyze the collected data. The data were presented as mean ± standard deviation (SD). Group comparisons were conducted using one-way analysis of variance (ANOVA) followed by Tukey multiple comparison tests. Unpaired t-tests were utilized to compare each group with the positive control group. A P-value of less than 0.05 was considered statistically significant.
Results
The results of the In vivo study
Influence of different azilsartan dosages on serum lipid profiles and atherogenic indices
Figure 1 illustrates the impact of graded concentration of azilsartan on the serum lipid profiles and atherogenic indices in the rat model challenged with ethanol. The administration of azilsartan at doses of 1 mg/kg and 10 mg/kg did not induce notable alterations in serum lipid profiles in comparison to the ethanol-treated group, which served as the positive control except for serum Very low density lipoprotein (VLDL) in which the level was significantly reduced by the low dose of azilsartan compared to the ethanol group with (p=0.0411; Figures 1A-E). Although the change in both the cardiac risk ratio and the atherogenic coefficient is not significant, azilsartan (10mg/kg) shows a significant escalation in the atherogenic index of plasma indicating an increase when compared to the positive control group.
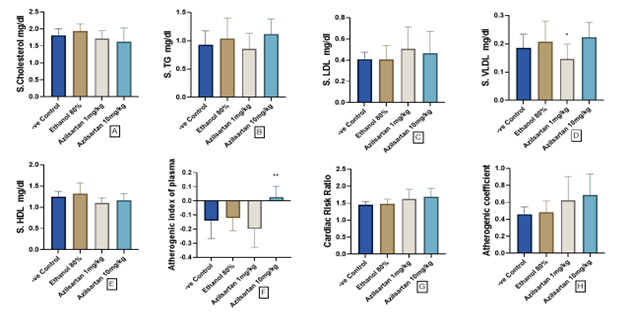 Figure 1. Effect of azilsartan on A) Cholesterol, B) Triglycerides (TG), C) Low density lipoprotein (LDL), D) Very low density lipoprotein (VLDL), E) High density lipoprotein (HDL), F) Atherogenic index, G) Cardiac risk ratio and H) Atherogenic coefficient. Values were presented as mean ± S.D (n= 8 animals in each group); values with (*) are significantly different from the positive control using ANOVA and post hoc test (* p<0.05), and (** p<0.01).
Figure 1. Effect of azilsartan on A) Cholesterol, B) Triglycerides (TG), C) Low density lipoprotein (LDL), D) Very low density lipoprotein (VLDL), E) High density lipoprotein (HDL), F) Atherogenic index, G) Cardiac risk ratio and H) Atherogenic coefficient. Values were presented as mean ± S.D (n= 8 animals in each group); values with (*) are significantly different from the positive control using ANOVA and post hoc test (* p<0.05), and (** p<0.01).
Influence of different azilsartan dosages on serum liver enzyme levels
The findings of the present investigation demonstrated a significant decrease in aspartate aminotransferase (AST) levels following the administration of 10 mg/kg azilsartan (p=0.0196), compared to the ethanol-treated cohort. This reduction was akin to that observed in the negative control cohorts (p=0.008) when juxtaposed against the positive control group (Figure 2A). Furthermore, regarding the impact on serum alanine transaminase (ALT) levels, a significant reduction was observed with the administration of azilsartan at a dosage of 10 mg/kg (p=0.0408) in contrast to the positive control group (Figure 2B).
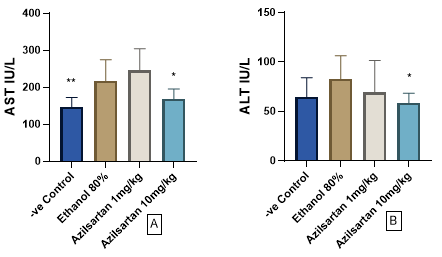
Figure 2. Effect of azilsartan on A) aspartate aminotransferase (AST), and B) alanine aminotransaminase (ALT) .Values were presented as mean ± S.D (n= 8 animals in each group); values with (*) are significantly different from the positive control using ANOVA and post hoc test (* p<0.05), and (** p<0.01).
Influence of different azilsartan dosages on levels of inflammatory and oxidative stress markers in the blood
The positive control group showed a sgnificant elevation in CRP levels compared to the negative control group (p<0.0001). Both 1 mg/kg and 10 mg/kg doses of azilsartan exhibited a significant decrease in CRP levels (p<0.0001) compared to the positive control group, similar to levels observed in the negative control group. Conversely, the ethanol-treated group displayed a significant rise in TNF-α serum levels compared to the negative control group (p<0.0001). However, administration of 10 mg/kg azilsartan resulted in a significant reduction in TNF-α levels (p=0.004) compared to the positive control group. Moreover, the neutrophil-to-lymphocyte ratio (NLR) notably increased in the ethanol-treated group compared to the negative control group (p=0.0097), with the high dose of azilsartan significantly attenuating this ratio compared to the ethanol group (p=0.023). Monocyte to lymphocyte ratio (MLR) significantly increased in the ethanol-treated group compared to the negative control group, (p=0.033), and 10 mg/kg of azilsartan decreased the ratio when compared to the ethanol group; however, it failed to achieve statistical significance. Additionally, no significant changes were observed in platelets to lymphocyte ratio (PLR), (p>0.05; Figure 3A-E).
In this study, the overall antioxidant capacity notably declined with ethanol administration in contrast to the control group without treatment, as indicated by a substantial decrease (p=0.002). However, the introduction of azilsartan at a dosage of 10 mg/kg significantly reversed this reduction (p<0.0001). Conversely, the application of 1 mg/kg azilsartan yield no significant difference compared to the ethanol-treated group (refer to Figure 4A). Moreover, the level of MDA, a marker of oxidative stress, significantly increased with ethanol exposure compared to the untreated control group (p<0.0001). Remarkably, both doses of azilsartan (1mg/kg and 10mg/kg) effectively mitigated this elevation, demonstrating significant reductions (p<0.001) and (p<0.0001) respectively, compared to the ethanol-treated group (Figure 3 F and G).
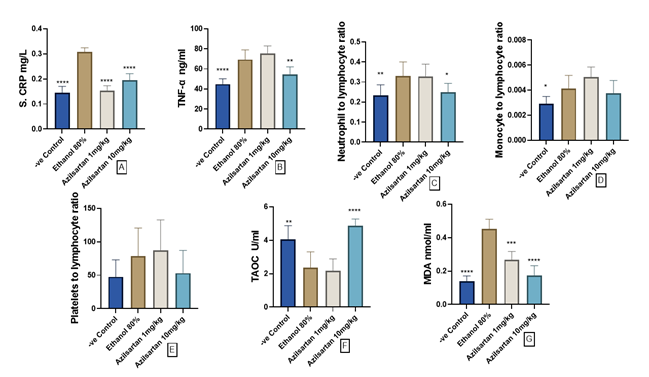
Figure 3. Effect of azilsartan on A) C-reactive protein (CRP), B) Tumor necrosis alpha (TNF-α), C) Neutrophil to lymphocyte ratio (NLR), D) Monocyte to lymphocyte ratio (MLR), E) Platelets to lymphocyte ratio (PLR), F) Total antioxidant capacity (TAOC), and G) Malodialdehyde (MDA). Values were presented as mean ± S.D (n= 8 animals in each group); values with (*) are significantly different from the positive control using ANOVA and post hoc test (* p<0.05), (** p<0.01), (*** p<0.001), and (**** p<0.0001).Influence of different doses of azilsartan on hematological markers
Table 1 displays the average values of hematologic parameters observed in both control and experimental groups following a 15-day administration of azilsartan. The data suggests that there were no notable variances in hemoglobin (Hb), hematocrit (HCT), red blood cell (RBC) count, or platelet count when compared to the control group. Conversely, a statistically significant increase was found in white blood cell (WBC) count in the control group compared to the ethanol-treated group (p=0.034).
Table 1. Effect of azilsartan on hematological markers in rats challenged with ethanol
| Parameter |
Negative control D.W.
1 ml (n=8) |
Ethanol 80%
1 ml (n=8) |
Azilsartan
1 mg/kg (n=8) |
Azilsartan
10 mg/kg (n=8) |
| Hb g/dl |
15.3 ± 1 |
15.7 ± 0.5 |
14.2 ± 1.9 |
15.1 ± 1.9 |
| Hct (%) |
49.2 ± 2.4 |
47.75 ± 1.7 |
43.6 ± 4.5 |
46.7 ± 5.2 |
| RBC (×109 cells/l) |
8 ± 0.7 |
8.1 ± 0.28 |
7.3 ± 0.6 |
7.8 ± 1.1 |
| WBC (×109 cells/l) |
18.5 ± 5.9* |
12.6 ± 3.7 |
16.9 ± 3.6 |
11.7 ± 3 |
| Platelet (×109 cells/l) |
642 ± 164 |
649 ± 117 |
779 ± 101 |
755 ± 264 |
Values are mean ± STD; N: number of animals; * significantly different from the ethanol-treated group (paired t-test, P<0.05); Hb: Hemoglobin; Hct: hematocrit; RBC: red blood cells; WBC: white blood cell
The results of molecular docking
In the present study, azilsartan's binding affinities against six important protein targets associated with heart and liver protection were studied in silico. Targets include AT1R, inflammatory markers (TNF-α, and CRP), oxidative stress (MDA), and liver enzymes (ALT, and AST). Co-crystallized structures of the protein targets were sourced from the Protein Data Bank (PDB IDs: 7T83, 7DOV, 1B09, 1Z9H, 3TCM, and 1AAM), respectively.
Molecular docking of azilsartan – AT1R interaction
Azilsartan's molecular docking results against the AT1R (PDB ID: 7T83), showed a good binding affinity of -8.3 kcal/mol, indicating that it has a favorable interaction profile with AT1R. As shown in Figure 4A, Azilsartan establishes two hydrogen bonds with important amino acid residues, ARG33 and SER55. The presence of the ethoxy group and the imidazole ring in azilsartan is likely to enhance the stability of its binding to the AT1R through the formation of hydrogen bonds. Significant hydrophobic interactions have been identified including pi-alkyl interactions with ARG33 and ARG99 due to the presence of aromatic rings in the azilsartan structure.
Molecular docking of azilsartan–inflammatory markers interaction
Azilsartan's molecular docking studies demonstrate that it has potential therapeutic effects against two inflammatory markers (TNF-α and CRP). It has a higher binding affinity for TNF-α than CRP, suggesting that it may alter inflammatory responses based on binding affinities and interaction patterns.
As observed in Figure 4B, the docking result shows that azilsartan and TNF-α have a binding affinity of -9.2 kcal/mol (PDB ID: 7DOV). An important interaction between Azilsartan and TNF-α, a crucial cytokine implicated in systemic inflammation, is suggested by this high binding affinity. The presence of carboxyl and ethoxy groups in azilsartan is crucial for establishing strong hydrogen bonding interactions with the amino acid residues ASN47 and GLN54 of TNF-α. These interactions enhance the binding affinity of ligand to TNF-α, thereby potentially improving its inhibitory effects on this pro-inflammatory cytokine. In addition, the 5-oxo-1,2,4-oxadiazole moiety and the aromatic rings in azilsartan play a critical role in establishing hydrophobic contacts through pi-alkyl and pi-sigma interactions with the amino acid residues ARG81 and VAL56.
The CRP-azilsartan complex formed five hydrophobic bonds which include alkyl, pi-alkyl, pi-sigma, pi-pi stacked, and pi-piTstacked with LEU43, VAL153, PRO12, LYS201, LEU204, VAL10, TYR40, and PHE199 through the interaction of methyl, aromatic rings, and imidazole ring, which are essential for hydrophobic interactions (Figure 4C). The binding affinity of this complex is indicated by a docking score of -8.1 kcal/mol, reflecting a good interaction between the ligand and the CRP protein.
Molecular docking of azilsartan–oxidative stress interaction
Azilsartan, a compound, has shown significant binding affinity against oxidative stress-related protein, MDA (PDB ID: 1Z9H).
Azilsartan's docking studies against MDA, produce a binding score of -8.4 kcal/mol, exhibiting a favorable binding affinity via both hydrogen and hydrophobic interactions, which are essential for the complex's stability. The complex formed two hydrogen bond interactions through the amino acid residues THR109 and TYR251, facilitated by the presence of the carboxylic acid functional group. The carboxylic acid group acts as a hydrogen bond donor and acceptor, enabling effective interactions with the hydroxyl group of THR109 and the carbonyl group of TYR251. Additionally, the complex exhibits five hydrophobic bonds with the amino acid residues CYS110, PRO111, PRO134, and ILE246 that are referred to for alkyl interactions (Figure 4D). Azilsartan's structure possesses aromatic rings as well as an imidazole ring, which facilitates these interactions.
Molecular docking of the interaction between azilsartan and liver enzymes
The molecular docking study of azilsartan against the liver enzymes ALT (PDB ID: 3TCM) and AST (PDB ID: 1AAM) revealed that azilsartan has a significantly higher binding affinity for ALT than for AST, indicating a stronger interaction that may affect its metabolic actions in the liver. The ALT-azilsartan complex exhibited a favorable docking score of -9.5 kcal/mol, demonstrating a strong inhibitory effect on the ALT receptor. The carboxylic acid group of azilsartan formed three hydrogen bonds with amino acid residues ALA148, SER149, and ARG308. Additionally, azilsartan established six strong hydrophobic interactions, including pi-alkyl, pi-pi stacked, pi-pi T stacked, and pi-cation bonds, which may enhance the stability of its binding to the target protein through interactions with the aromatic rings, imidazole ring, and 5-oxo-1,2,4-oxadiazole alongside amino acid residues ARG22, VAL260, TYR19, TYR174, and SER298 (Figure 4E).
Azilsartan exhibits binding affinities that target the AST receptor, with docking scores of -8.8 kcal/mol. These binding interactions enable the formation of hydrogen bonding; the presence of hydroxyl group is necessary for mediating this hydrogen bond interaction with GLN321. Furthermore, aromatic rings, imidazole rings, and 5-oxo-1,2,4-oxadiazole play important roles in hydrophobic interactions with amino acid residues PRO56, LEU58, ILE318, MET326, ARG329, ASP50, GLU322, and ASP325 (Figure 4F).

Figure 4. A) The binding mode of Azilsartan (Turquoise) at the pocket of the 7T83 and the 2D intermolecular binding interactions. B) Azilsartan (purple) binding pattern in the 7DOV pocket site, as well as 2D intermolecular binding interactions. C) Azilsartan (Green) binds to a specific pocket within the CRP and the 2D intermolecular binding interactions. D) Azilsartan (Grey) binding mode in the pocket site of the 1Z9H, and the 2D intermolecular binding interactions. E) The binding mode of azilsartan (Orange) at the pocket site of the 3TCM and the 2D intermolecular binding interactions. F) The binding mode of azilsartan (Yellow) at the pocket site of the 1AAM and the 2D intermolecular binding interactions.
Discussion
In vivo study
Several mechanisms are involved in alcohol-induced hepatic and cardiac toxicity. The liver is the primary organ for alcohol metabolism producing harmful byproducts that damage liver cells. Persistent alcohol intake may result in liver conditions such as fatty liver, alcoholic hepatitis, and cirrhosis [24]. Cardiomyopathy is one of the well-known mechanisms of alcohol-induced cardiac toxicity [25]. Therefore, it is imperative to establish initiatives aimed at preventing, detecting early, and providing tailored treatment for the pertinent ailment [26]. Additional factors such as variations in genetic makeup, gender distinctions, concurrent use of substances (e.g., tobacco or cocaine), as well as the presence of other cardiovascular risk factors such as hypertension and diabetes, have the potential to impact and exacerbate the progression of arrhythmogenic cardiomyopathy in individuals, thereby emphasizing the need for comprehensive care strategies [27]. The breakdown of ethanol through the alcohol dehydrogenase (ADH) pathway yields acetaldehyde and nicotinamide adenine dinucleotide (NADH) reduced form. The detrimental impacts of ethanol largely stem from alterations in the body's redox balance, characterized by elevated levels of NADH and depleted levels of nicotinamide adenine dinucleotide (NAD+). This shift in redox equilibrium, primarily driven by the ADH pathway, is associated with various metabolic disorders such as hyperlipidemia [28]. In the present study, a non-significant elevation in lipid profiles was observed following ethanol administration, while a low dose of azilsartan effectively reduced triglyceride levels. Moreover, evidence shows that atherogenic indices are important in predicting cardiovascular risk, as the value increases, so does the risk of cardiovascular disease, and conversely, as the value decreases, the risk also decreases [29,30]. To the best of our knowledge, this study is the first to link azilsartan intake with lipid cardiovascular indices. A low dose of azilsartan in the current study resulted in a significant reduction in the AIP. Azilsartan has been found in previous studies to attenuate atherosclerosis [31,32]. Furthermore, azilsartan has been shown to restore nitric oxide levels and thereby improve endothelium dysfunction [31]. The increased liver enzyme activities are generally considered to be a sign of hepatic diseases. The administration of the azilsartan ameliorated the levels of ALT and AST with simultaneous reductions in hepatic lipid peroxide levels, indicating a clear cytoprotective effect against alcohol-induced oxidative damage to liver tissues. The inflammatory biomarkers, including CRP and TNF-α increased in response to ethanol administration. Ethanol triggers the activation of the NF-KB pathway, prompting the generation of several proinflammatory cytokines such as TNF-α. Consequently, this cascade initiates the expression of inflammatory indicators such as CRP. C-reactive protein, synthesized primarily by hepatocytes, is a circular pentameric protein whose concentration increases during inflammation. Elevated levels of CRP and TNF-α serve as indicators of alcohol-induced liver damage [33,34]. Other novel inflammatory markers, such as NLR and PLR, have recently been involved in the toxicity associated with alcohol-induced liver injury [35]. Ethanol administration elevated these ratios, although the increase was not statistically significant; a high dose of azilsartan significantly attenuated these ratios and the manifestation of other inflammatory biomarkers such as CRP and TNF-α. According to a study by Kajiya et al. in 2011, a cellular test revealed that azilsartan prevented the activation of mitogen-activated protein kinases in vascular smooth muscle cells triggered by angiotensin II (AII), even after the drug was washed out with a delay [36]. This effect is likely due to azilsartan's inherent high affinity and strong binding properties [37]. Studies hypothesized that azilsartan demonstrated pleiotropic effects by potently boosting the expression of PPAR-γ while decreasing TNF-α production [38]. In line with the current findings, Hye Khan et al. (2014a) explained the protective effects of azilsartan in Zucker diabetic fatty rats stem from its ability to enhance glucose regulation, promote better balance within blood vessels, and reduce the levels of oxidative stress and inflammation [39]. Furthermore, azilsartan was shown to recover endothelium function in animal studies, as in diabetic mice normalization of eNOS function, along with the reduction of inflammation and oxidative stress, was observed [31]. A proposed mechanism of the pleiotropic effects of azilsartan could be related to the activation of PPAR-γ, an intracellular receptor that regulates lipid and glucose metabolism [40]. TAOC represents a crucial marker reflecting the body's antioxidant prowess, intimately linked with non-enzymatic antioxidant defense mechanisms. Alcohol administration led to a notable decline in serum TAOC levels compared to the control group. However, pre-treatment of rats with azilsartan, at doses of 1 and 10 mg/kg prior to ethanol exposure, effectively replenished TAOC levels. Notably, there was a dose-dependent enhancement, with the highest dosage of azilsartan yielding the most substantial increase, akin to the standard group, indicating reinforcement of the rats' non-enzymatic antioxidant defense systems. MDA, an indirect marker of oxidative stress and a byproduct of lipid peroxidation causing cellular membrane damage was restored to baseline levels with the higher dosage of azilsartan. A study connected the protective antioxidant effect of azilsartan on brain endothelial cell dysfunction from oxidative stress to the activation of the PPAR-γ pathway [32]. Azilsartan has recently been proven to have a gastroprotective effect through increasing antioxidant capacity and attenuating inflammatory response [41]. Moreover, the present study revealed that the hematologic parameters were not significantly altered by each of the treatment groups, except for the WBC level which decreased significantly in the ethanol-treated group [42]. These findings suggest that azilsartan has no negative effect on the hematological markers [43].
Molecular docking
Molecular docking studies spotlight azilsartan's binding affinities and processes, which helps to understand better how the drug interacts with cardiac and hepatic receptors and its pocket site. The docking results showed that azilsartan interacted with the proteins 7T83, 7DOV, 1B09, 1Z9H, 3TCM, and 1AAM, confirming their inhibitory effects on targets. The affinity values ranged from -9.5 to -8.1 kcal/mol, with more negative values indicating a stronger binding affinity and a greater inhibitory effect or blockage against the target proteins. The obtained results, which are consistent with experimental findings, are enhanced by the combined results of in vivo and computational studies, which are supported by biochemical evidence.
As illustrated in figures 5-10, hydrogen bonding interactions play a significant role in the binding ligand to various target proteins through functional groups such as carboxyl, hydroxyl, ethoxy, 5-oxo-1,2,4-oxadiazole, and imidazole rings, with the exception of protein 1B09. In addition, hydrophobic interactions contribute substantially to the binding of the drug to the target proteins via functional groups, including aromatic rings, alkyl groups, imidazole, and 5-oxo-1,2,4-oxadiazole. These interactions significantly reduce blood pressure in individuals with hypertension and have been shown to confer cardioprotective effects, inhibit inflammation, mitigate oxidative stress, and reduce liver enzyme levels. Therefore, azilsartan contains various functional groups, including both electron-donating and electron-withdrawing groups, which facilitate strong binding to target proteins and have a range of pharmacological effects.
Studies have shown that the structure of azilsartan differs from that of other ARBs due to the presence of a unique moiety: a 5-oxo-1,2,4-oxadiazole ring, which replaces the tetrazole ring found in candesartan, valsartan, olmesartan, losartan, and irbesartan. It has been proposed that even minor variations in the molecular structures of ARBs can result in significant differences in their capacity to interact with the AT1 receptor. Likewise, small structural alterations in ligands for other G-protein-coupled receptors might result in different pharmacological effects [37, 44, 45].
The finding of a study by Miura et al. revealed that azilsartan and candesartan bind to the same AT1 receptor sites, with stronger hydrogen bonding between the oxadiazole of azilsartan and Gln257 than that between the tetrazole of candesartan and Gln257. This interaction reduces blood pressure in hypertension patients and has cardioprotective effects. It is suggested that azilsartan's unique binding behavior, attributed to its 5-oxo-1,2,4-oxadiazole moiety, may explain its superior BP-lowering efficacy compared to candesartan and other ARBs [46].
Conclusions
The findings of the current study demonstrate the hepatoprotective and cardioprotective activity of azilsartan against ethanol-evoked damage both in vivo and in silico studies. The suggested pathways involve ameliorating oxidative stress and inflammation. These findings may indicate the effectiveness of azilsartan for other therapeutic purposes. Nonetheless, further experimental and clinical investigations are necessary to confirm these preliminary results in clinical settings.
Data Access and Responsibility
The data are available upon request.
Ethical Considerations
The research procedures adhered to the guidelines outlined by the Institutional Animal Ethics Committee and were approved by the Ethical Committee of the University of Sulaimani, College of Pharmacy (Certificate No. PH34-21 on October 20, 2023).
Authors' Contributions
Renas Raouf Hama Amin: Methodology, Software, Validation, Investigation, Resources, Funding Acquisition, and Visualization. Tavga Ahmed Aziz: Conceptualization, Investigation, Project administration, Formal analysis, Supervision, Writing – review & editing the first and final draft. Narmin Hamaamin Hussen: Methodology, Software, Validation, Investigation, Resources, and Funding Acquisition. Zheen Aorahman Ahmed: Methodology, Validation, Visualization, and Project administration.
Acknowledgement
The authors thank the College of Pharmacy and the University of Sulaimani for providing the facilities and support needed for this project.
Conflict of Interests
The authors declare no conflicts of interest.
Funding
This study received no fundings.
References
- Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4(3):1201. [DOI: 10.1002/cphy.c130040]
- Husain A, Graham RM. Drugs, enzymes and receptors of the renin-angiotensin system: celebrating a century of discovery. CRC Press; 2000 Feb 23. [Link]
- Tziomalos K, Athyros VG, Paschos P, Karagiannis A. Nonalcoholic fatty liver disease and statins. Metabolism. 2015;64(10):1215-23. [DOI: 10.1016/j.metabol.2015.07.003] [PMID: 26234727]
- Fyhrquist F, Saijonmaa O. Renin‐angiotensin system revisited. J Intern Med. 2008;264(3):224-36. [DOI: 10.1111/j.1365-2796.2008.01981.x] [PMID: 18793332]
- Leung PS. The physiology of a local renin–angiotensin system in the pancreas. J physiol. 2007;580(1):31-7. [DOI: 10.1113/jphysiol.2006.126193] [PMID: 17218353]
- Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, Solé M, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125(1):117-25. [DOI: 10.1016/s0016-5085(03)00695-4]
- Nabeshima Y, Tazuma S, Kanno K, Hyogo H, Iwai M, Horiuchi M, et al. Anti-fibrogenic function of angiotensin II type 2 receptor in CCl4-induced liver fibrosis. Biochem Biophys Res Commun. 2006;346(3):658-64. [DOI: 10.1016/j.bbrc.2006.05.183]
- Santos RA, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M. The renin-angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol. 2019;316(5):H958-70. [DOI: 10.1152/ajpheart.00723.2018] [PMID: 30707614]
- Namsolleck P, Recarti C, Foulquier S, Steckelings UM, Unger T. AT 2 receptor and tissue injury: therapeutic implications. Curr Hypertens Rep. 2014;16:416. [DOI: 10.1007/s11906-013-0416-6] [PMID: 24414230]
- Bruce E, Shenoy V, Rathinasabapathy A, Espejo A, Horowitz A, Oswalt A, et al. Selective activation of angiotensin AT 2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br J Pharmacol. 2015;172(9):2219-31. [DOI: 10.1111/bph.13044] [PMID: 25522140]
- Gao J, Zucker IH, Gao L. Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure. Am J Hypertens. 2014;27(10):1248-56. [DOI: 10.1093/ajh/hpu044] [PMID: 24687998]
- de Kloet AD, Steckelings UM, Sumners C. Protective angiotensin type 2 receptors in the brain and hypertension. Curr Hypertens Rep. 2017;19:46. [DOI: 10.1007/s11906-017-0746-x] [PMID: 28488048]
- Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens. 1999;12(S9):205S-13S. [DOI: 10.1016/s0895-7061(99)00103-x]
- Miura SI, Matsuo Y, Nakayama A, Tomita S, Suematsu Y, Saku K. Ability of the new AT1 receptor blocker azilsartan to block angiotensin II-induced AT1 receptor activation after wash-out. J Renin-Angiotensin-Aldosterone Sys. 2014;15(1):7-12. [DOI: 10.1177/1470320313482170]
- Li W, Wang C, Zhang D, Zeng K, Xiao S, Chen F, et al. Azilsartan ameliorates ox-LDL-induced endothelial dysfunction via promoting the expression of KLF2. Aging (Albany NY). 2021;13(9):12996-3005. [DOI: 10.18632/aging.202973]
- Gupta V, Dhull DK, Joshi J, Kaur S, Kumar A. Neuroprotective potential of azilsartan against cerebral ischemic injury: Possible involvement of mitochondrial mechanisms. Neurochem Int. 2020;132:104604. [DOI: 10.1016/j.neuint.2019.104604]
- Hussain SA, Utba RM, Assumaidaee AM. Effects of azilsartan, aliskiren or their combination on high fat diet-induced non-alcoholic liver disease model in rats. Med Arch. 2017;71(4):251-5. [DOI: 10.5455/medarh.2017.71.251-255] [PMID: 28974844]
- Mshelia HS, Karumi Y, Dibal NI. Therapeutic effect of Momordica balsamina leaf extract on ethanol-induced gastric ulcer in Wistar rats. Ann Res Hosp. 2017;1(1). [DOI:10.21037/arh.2017.04.03]
- de Araújo AA, Varela H, de Medeiros CA, de Castro Brito GA, de Lima KC, de Moura LM, et al. Azilsartan reduced TNF-α and IL-1β levels, increased IL-10 levels and upregulated VEGF, FGF, KGF, and TGF-α in an oral mucositis model. PloS One. 2015;10(2):e0116799. [DOI: 10.1371/journal.pone.0116799] [PMID: 25689279]
- Aluko EO, Omobowale TO, Oyagbemi AA, Adejumobi OA, Ajibade TO, Fasanmade AA. Reduction in nitric oxide bioavailability shifts serum lipid content towards atherogenic lipoprotein in rats. Biomed Pharmacother. 2018;101:792-7. [DOI: 10.1016/j.biopha.2018.03.001]
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11(5):905-19. [DOI: 10.1038/nprot.2016.051] [PMID: 27077332]
- Burley SK, Berman HM, Duarte JM, Feng Z, Flatt JW, Hudson BP, et al. Protein data bank: a comprehensive review of 3D structure holdings and worldwide utilization by researchers, educators, and students. Biomolecules. 2022;12(10):1425. [DOI: 10.3390/biom12101425] [PMID: 36291635]
- Terefe EM, Ghosh A. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of phytochemicals isolated from Croton dichogamus against the HIV-1 reverse transcriptase. Bioinform Biol Insights. 2022;16. [DOI: 10.1177/11779322221125605] [PMID: 36185760]
- Fernández-Solà J. The effects of ethanol on the heart: alcoholic cardiomyopathy. Nutrients. 2020;12(2):572. [DOI: 10.3390/nu12020572]
- Moure-Rodriguez L, Carbia C, Lopez-Caneda E, Corral Varela M, Cadaveira F, Caamaño-Isorna F. Trends in alcohol use among young people according to the pattern of consumption on starting university: A 9-year follow-up study. PLoS One. 2018;13(4):e0193741. [DOI: 10.1371/journal.pone.0193741] [PMID: 29630657]
- Bagnardi V, Sorini E, Disalvatore D, Assi V, Corrao G, De Stefani R, et al. ‘Alcohol, less is better’project: outcomes of an Italian community‐based prevention programme on reducing per‐capita alcohol consumption. Addiction. 2011;106(1):102-10. [DOI: 10.1111/j.1360-0443.2010.03105.x]
- Steiner JL, Lang CH. Etiology of alcoholic cardiomyopathy: mitochondria, oxidative stress and apoptosis. Int J Biochem Cell Biol. 2017;89:125-35. [DOI:10.1016/j.biocel.2017.06.009]
- Govindan S, Jayabal A, Shanmugam J, Ramani P. Antioxidant and hepatoprotective effects of Hypsizygus ulmarius polysaccharide on alcoholic liver injury in rats. Food Sci Hum Wellness. 2021;10(4):523-35. [DOI:10.1016/j.fshw.2021.04.015]
- Ahmad MN. The effect of lentil on cholesterol-induced changes of serum lipid cardiovascular indexes in rats. Prog Nutr. 2017;19(1):48-56. [DOI:10.2375/pn.v19i1.4856]
- Yu L, Gao R, Song X, Li X, Zhu J. Cardio-protective and anti-atherosclerosis effect of crocetin on vitamin D3 and HFD-induced atherosclerosis in rats. J Oleo Sci. 2021;70(10):1447-59. [DOI: 10.5650/jos.ess21168] [PMID: 34615830]
- Matsumoto S. Shimabukuro M1, Fukuda D, Soeki T, Yamakawa K, Masuzaki H, et al. Azilsartan, an angiotensin II type 1 receptor blocker, restores endothelial function by reducing vascular inflammation and by increasing the phosphorylation ratio Ser1177/Thr497 of endothelial nitric oxide synthase in diabetic mice. Cardiovasc Diabetol. 2014;13:30. [DOI: 10.1186/1475-2840-13-30] [PMID: 24485356]
- Liu H, Mao P, Wang J, Wang T, Xie CH. Azilsartan, an angiotensin II type 1 receptor blocker, attenuates tert-butyl hydroperoxide-induced endothelial cell injury through inhibition of mitochondrial dysfunction and anti-inflammatory activity. Neurochem Int. 2016;94:48-56. [DOI: 10.1016/j.neuint.2016.02.005]
- Lin CJ, Chiu CC, Chen YC, Chen ML, Hsu TC, Tzang BS. Taurine attenuates hepatic inflammation in chronic alcohol-fed rats through inhibition of TLR4/MyD88 signaling. J Med Food. 2015;18(12):1291-8. [DOI: 10.1089/jmf.2014.3408] [PMID: 26090712]
- OGO O, Obochi G, Ikwebe J, Ogli S, Amali O. Biochemical and haematological changes associated with ethanol-induced gastric lesions in Wistar rats. Int J Toxicol Appl Pharmacol. 2016;6(1):7-11. [Link]
- Khan RS, Lalor PF, Thursz M, Newsome P. The role of neutrophils in alcohol-related hepatitis. J Hepatol. 2023;79(4):1037-48. [DOI: 10.1016/j.jhep.2023.05.017]
- Kajiya T, Ho C, Wang J, Vilardi R, Kurtz TW. Molecular and cellular effects of azilsartan: a new generation angiotensin II receptor blocker. J //hypertens. 2011;29(12):2476-83. [DOI: 10.1097/HJH.0b013e32834c46fd] [PMID: 21986624]
- Ojima M, Igata H, Tanaka M, Sakamoto H, Kuroita T, Kohara Y, et al. In vitro antagonistic properties of a new angiotensin type 1 receptor blocker, azilsartan, in receptor binding and function studies. J Pharmacol Exp Ther. 2011;336(3):801-8. [DOI: 10.1124/jpet.110.176636]
- Iwai M, Chen R, Imura Y, Horiuchi M. TAK-536, a new AT1 receptor blocker, improves glucose intolerance and adipocyte differentiation. Am J Hypertens. 2007;20(5):579-86. [DOI: 10.1016/j.amjhyper.2006.12.010]
- Khan MA, Neckář J, Haines J, Imig JD. Azilsartan improves glycemic status and reduces kidney damage in zucker diabetic fatty rats. Am J Hypertens. 2014;27(8):1087-95. [DOI: 10.1093/ajh/hpu016]
- Kurtz TW, Pravenec M. Molecule-specific effects of angiotensin ii–receptor blockers independent of the renin–angiotensin system. Am J Hypertens. 2008;21(8):852-9. [DOI: 10.1038/ajh.2008.202]
- Hama Amin RR, Aziz TA. Gastroprotective effect of Azilsartan through ameliorating oxidative stress, inflammation, and restoring hydroxyproline, and gastrin levels in ethanol-induced gastric ulcer. J Inflamm Res. 2022;15:2911-23. [DOI: 10.2147/JIR.S365090] [PMID: 35592072]
- Nakanishi N, Yoshida H, Okamoto M, Matsuo Y, Suzuki K, Tatara K. Association of alcohol consumption with white blood cell count: a study of Japanese male office workers. J Intern Med. 2003;253(3):367-74. [DOI: 10.1046/j.1365-2796.2003.01112.x]
- Raheem NM, Mohammed Ali Mahmood N. Azilsartan suppresses the antiapoptotic biomarker and pro-inflammatory cytokines in rat model of cisplatin-induced retinal and optic nerve toxicity. Hum Exp Toxicol. 2023. [DOI: 10.1177/09603271231155092]
- Kurtz TW, Kajiya T. Differential pharmacology and benefit/risk of azilsartan compared to other sartans. Vasc Health Risk Manag. 2012;8:133-43. [DOI: 10.2147/VHRM.S22595] [PMID: 22399858]
- Fujino M, Miura S, Kiya Y, Tominaga Y, Matsuo Y, Karnik SS, et al . A small difference in the molecular structure of angiotensin II receptor blockers induces AT1 receptor-dependent and -independent beneficial effects. Hypertens Res. 2010;33:1044–52. [DOI: 10.1038/hr.2010.135] [PMID: 20668453]
- Miura S, Okabe A, Matsuo Y, Karnik SS, Saku K. Unique binding behavior of the recently approved angiotensin II receptor blocker azilsartan compared with that of candesartan. Hypertens Res. 2013;36(2):134-9. [DOI: 10.1038/hr.2012.147] [PMID: 23034464]









